
CASE REPORT
Chronic Granulomatous Disease Revealed by Multiple Cystic and Pseudo-tumoral Liver Lesions: One Case Report
Ibtissam Khattou1, *, Imane Ait Sab1, Noureddine Rada1, Aicha Bourrahouat1, Btissam Zouita2, Dounia Basraoui2, Hicham Jalal2, Mohamed Sbihi1
Article Information

Identifiers and Pagination:
Year: 2018Volume: 10
First Page: 151
Last Page: 155
Publisher ID: TOIDJ-10-151
DOI: 10.2174/1874279301810010151
Article History:
Received Date: 4/8/2018Revision Received Date: 3/10/2018
Acceptance Date: 10/10/2018
Electronic publication date: 24/10/2018
Collection year: 2018

open-access license: This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International Public License (CC-BY 4.0), a copy of which is available at: https://creativecommons.org/licenses/by/4.0/legalcode. This license permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Chronic Granulomatous Disease (CGD) is a rare immunodeficiency disease described as a lack of destruction of bacteria and fungi phagocytes by neutrophils and macrophages, it is related to an abnormality of NADPH oxidase, a free radical producer of oxygen. The most common aspect of CGD at the age of one year, is an infection of the skin or bone by two bacteria called staphylococcus aureus and serratia marcescens. In this article, the authors report a case of CGD revealed by multiple cystic and pseudo-tumoral liver lesions discovered during prolonged fever.
1. INTRODUCTION
Chronic Granulomatous Disease (CGD) is a rare immunodeficiency disease, secondary to a defect in the oxidative metabolism of phagocytes. The particularity of this article is to report a rare case of immune deficiency, revealed by an unusual radiological aspect. We report a case of CGD that is revealed by multiple cystic and pseudo-tumoral liver lesions.
2. CASE REPORT
The case was 2-year and 2-month-old female child, who had consanguineous parents, and having a history of repeated respiratory infections. She was admitted to our department for prolonged fever with deterioration of the general state, and in whom, the clinical examination found a failure to thrive (weight; 10kg (<-3SD) and height 78cm (<-3SD)), with nodular hepatomegaly at abdominal palpation.
The biological assessment showed hyperleukocytosis at 23310 elements/mm3 predominantly neutrophilic at 16600 elements/mm3. C-reactive protein (CRP) was elevated to 214 mg / l.
Abdominal ultrasound showed hepatic lesion taking the whole left liver measuring 81 * 57mm poorly limited with heterogeneous echo-structure, no flow in Doppler with the existence of another hepatic lesion at segment 4 measuring 22 * 16mm hyperechoic center surrounded by a hypoechoic halo and ill-defined borders.
Thoraco-abdominopelvic CT revealed a locally infiltrated left liver lesion involving the right hepatic artery associated with multifocal cystic lesions at the liver (Figs. 1A, B and 2), right thoracic wall lesion, and left excavated pulmonary lesion. These images indicate secondary locations (Figs. 3 and 4).
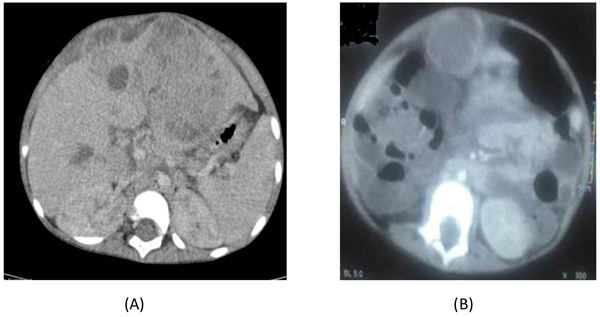 |
Fig. (1). (A) multi-leaf formation with thick wall, centered by a vessel, pseudo-tumor aspect (B) Infiltrating the hilar bifurcation responsible for portal thrombosis. |
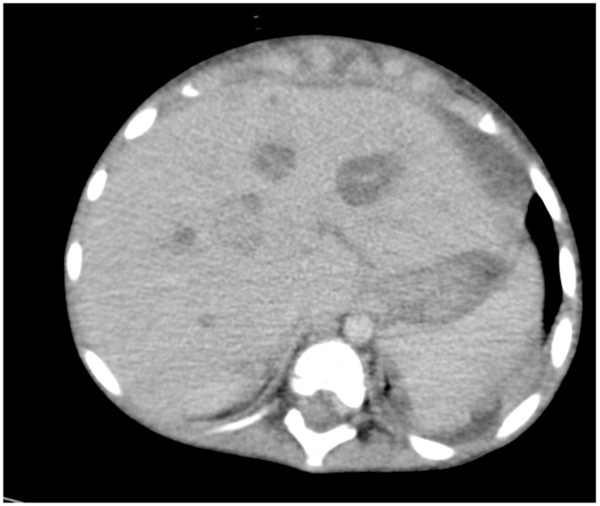 |
Fig. (2). Multiple Cystic and Liver Tissue lesions. |
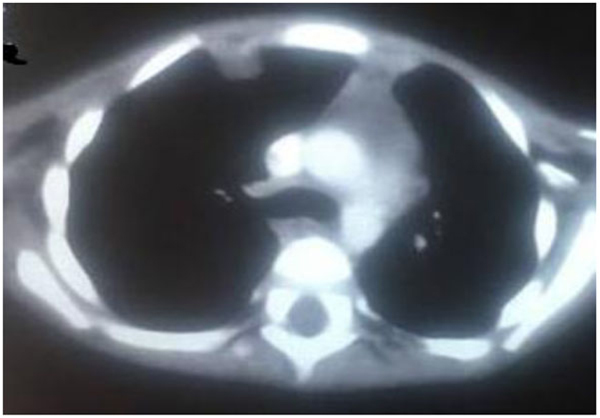 |
Fig. (3). Liquid thoracic wall mass with thick walls. |
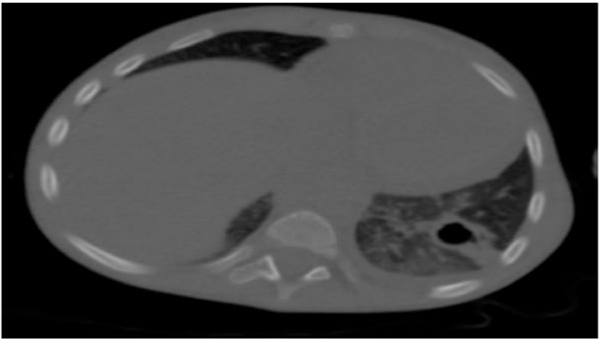 |
Fig. (4). Excavated parenchymal condensation. |
The dosage of alpha-fetoprotein was normal. A percutaneous liver biopsy was done but it was inconclusive which led to a surgical biopsy. The histopathological study showed a liver abscess without signs of malignancy and no isolate was identified. The patient was treated with tri-antibiotic therapy (Ceftriaxone + Gentamicin + Metronidazole). The lesion evolved toward the constitution of an abscess in front of the operative wound which was drained and the bacteriological study of the pus isolated Serratia marcescens.
Faced the hepatic abscess and the isolated bacteria, the diagnosis of immunodeficiency was suggested. An HIV serology was performed which was negative, the dosage of immunoglobulin and lymphocyte was normal. The exploration of oxidative burst of PNN (Neutrophil polynuclear) has shown a lack of response from the PNN (Dihydrorhodamine test).
The diagnosis of chronic septic granulomatous disease was retained and the patient was made to experience under anti-microproteinase basically trimethoprim-sulfamethoxazole with a good progress.
3. DISCUSSION
Chronic Granulomatous Disease (CGD) is a rare, primitive immune deficiency of genetic origin; the incidence is estimated to be between 1/200,000 and 1/250,000 births in the United States [1], it is secondary to a defect in the oxidative metabolism of neutrophils, monocytes, macrophages and eosinophils [2]. It is revealed by recurrent bacterial and fungal infections and by the development of granulomas with multi-visceral involvement. CGD usually occurs before the age of five, but it can be diagnosed in adulthood [3]. The favorite sites are lungs (80%), skin (60-70%), lymph nodes (75%), liver and digestive tract (32%) [4].Chronic inflammatory signs, particularly hepatic granulomatous infiltration responsible for hepatomegaly reported in 80-100% of cases [5], are among the cardinal signs of the disease. The diagnosis is based on the exploration of granulocyte bactericide mainly by an easy reduction test; the Nitroblue Tetrazolium (NBT) which remains the screening test of choice for this pathology.It is based on the activation of NADPH granulocyte oxidase by soluble agents, the Phorbol Myristate Acetate (PMA) in the presence of NBT. But the current trend is to develop the measurement of the fluorescence of dihydrorhodamine (DHR) in the presence of toxic derivatives of oxygen (ROS) [6, 7].The genetic study allows determining the CGD original mutation which will guide any possible familial study, but also it makes it possible proposing genetic counseling to the parents, as the possibility of an antenatal diagnosis during future pregnancy [6].
Children with CGD should receive anti-aspergilla and antibacterial chemoprophylaxis with trimethoprim-sulfamethoxazole (TMP-SMX), which is the most commonly used antibiotic because it has a good spectrum of activity on microorganisms faced in the CGD.TMP-SMX is lipophilic (therefore concentrated in the cell) and well tolerated, since it does not affect the intestinal flora [6]. Thanks to the natural evolution (reduction of infectious episodes with age) and therapeutic progress, especially of anti-infectious prophylaxis, patients diagnosed in childhood reach adulthood and can lead normal personal and professional life [8].
Bone marrow allograft, the only curative treatment for CGD, is only possible when there is a compatible HLA donor. Gene therapy using a vector (retrovirus or adenovirus) is a potential future treatment [9] as had been shown in the treatment of severe combined immunodeficiency [10].
CONCLUSION
The CGD is a rare primary immunodeficiency, the evolution of which under antibioprophylaxis is generally favorable; it should be suggested before any deep abscess in child and before any infection with unusual germs such as Burkholderia cepacia, Serratia marcescens, aspergillus and Nocardia.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
No animals/humans were used for studies that are the basis of this report.
CONSENT FOR PUBLICATION
A written informed consent was obtained from the parents.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
Declared none.





